ESM-2 (evolutionary-scale prediction of atomic level protein structure with a language model)
Highlights of the ESM-2 Paper
• Infer structure directly from primary sequence using LLM
• LM leverages the evolutionary patterns captured in the LM to produce atomic level predictions
• Order of magnitude faster (60x) in high res structure prediction
• Present a ESM metagenomic atlas (structural characterization of of more than 617 million$^\ddagger$ metagenomic proteins$^\dagger$)
1. Introduction
1.1 - Structure and Function is Hidden in Sequences.
Biological properties of proteins influence which position/(s) in its sequence can undergo mutations. Therefore, based on these observations, we can define types of evolutionary functions that has happened such as coevolution and conservation of amino acids. These observations can lead to infer properties regarding the function and structure of proteins.$^\star$
Usually we rely on aligning sequences before we can draw conclusions into the function and structure. This intermediate representation known as multiple sequence alignment (MSA) has a high time complexity as we have to 1) search for related sequences first$^\star$, and 2) align them.
What if we can get rid of this intermediate representation? That's one aspect this paper accomplishes.
1.2 - Large language models (LLMs)
Historically LMs were pretrained by using techniques such as predicting the next word in a sentence. But Devlin et al. [BERT] showed that just by masking some words in the input and trying to predict it ("masked language model objective - MLM") is a better pretraining strategy $^\star$.
1.3 - Contributions
Inspired by this widely adopted strategy, the authors of this paper hypothesise that filling missing amino acids might result in learning things which are valuable enough to infer the structure. Thus, they scale protein language models from 8 million parameters upto 15 billion parameters. Doing so reveals the following,
- Enable of atomic level structure prediction directly from sequence.
- strong correlation in perplexity and accuracy (of structure prediction)
- 60x speed improvement on inference
- No need for search process of related sequences
Because of this one to two orders of speed improvement and the fact that MSA is not needed, they expand structure prediction to metagenomic proteins which is much greater in extent and diverse as well. Therefore, in summary they,
- Predict structures for all sequences (over 617M) in MGnify90 $\dagger$
- Out of 617M proteins, 225M structures have high confidence.
- Out of high confidence ones 76.8% are disjoint from the UniRef90 dataset by atleast 90% of sequence identity.
- 12.6% have no experimental groundtruth.
- Out of 617M proteins, 225M structures have high confidence.
2. Method
2.1 - How does structure emerge from LM trained on sequences?
ESM-2 language model is trained with ~65 million unique sequences $^\bigstar$. Because of the MLM objective, we ask the model to predict missing pieces (amino acids) of the sequences using the neighbouring amino acid context. Therefore, the assumption is that the model needs to learn inter-dependencies of amino acids. In previous work [1] and [2], it was shown$^\dagger$ that transformer models trained with MLM on protein sequences develop attention patterns which corresponds to the residue-residue contact map.
After training the LM, in order to compute the contact map from attention patterns, the authors use the approach in [2], where they use logistic regression to identify contacts as follows.
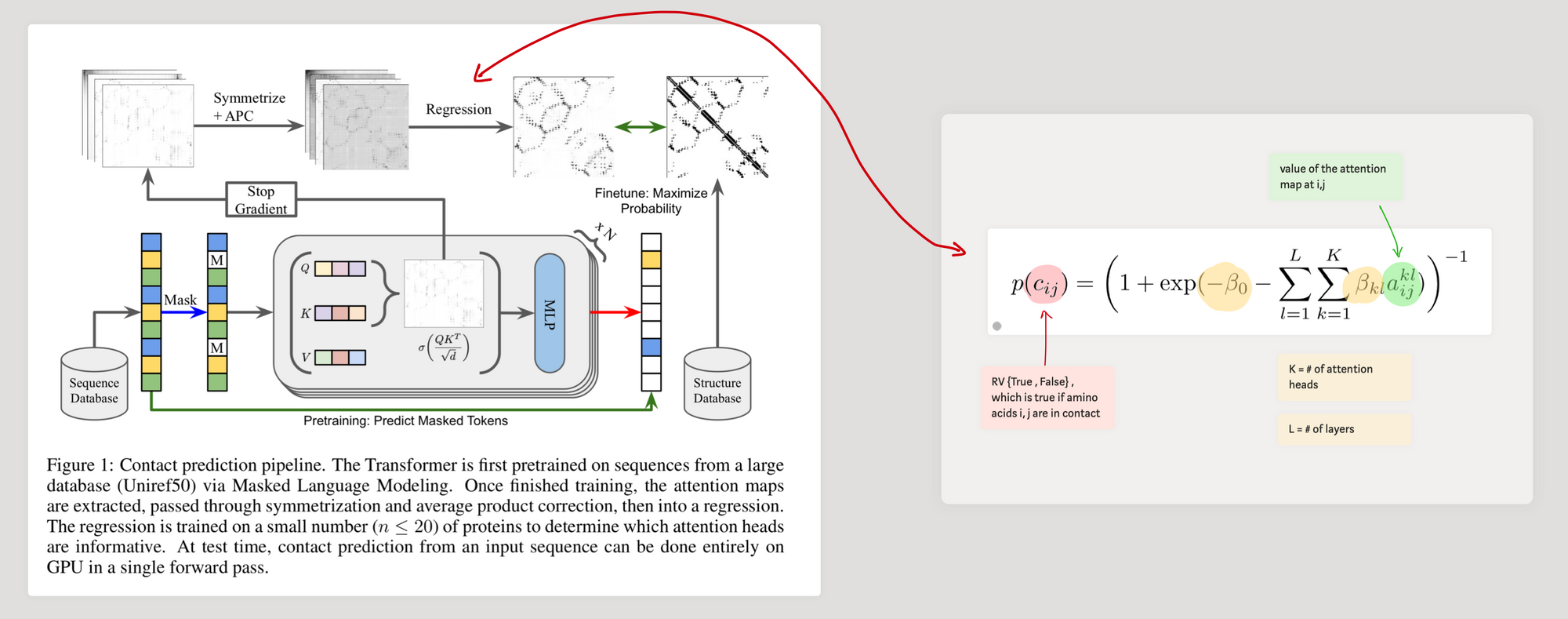
2.2 - Ok, what about atomic level structure? (Enter ESM-Fold)
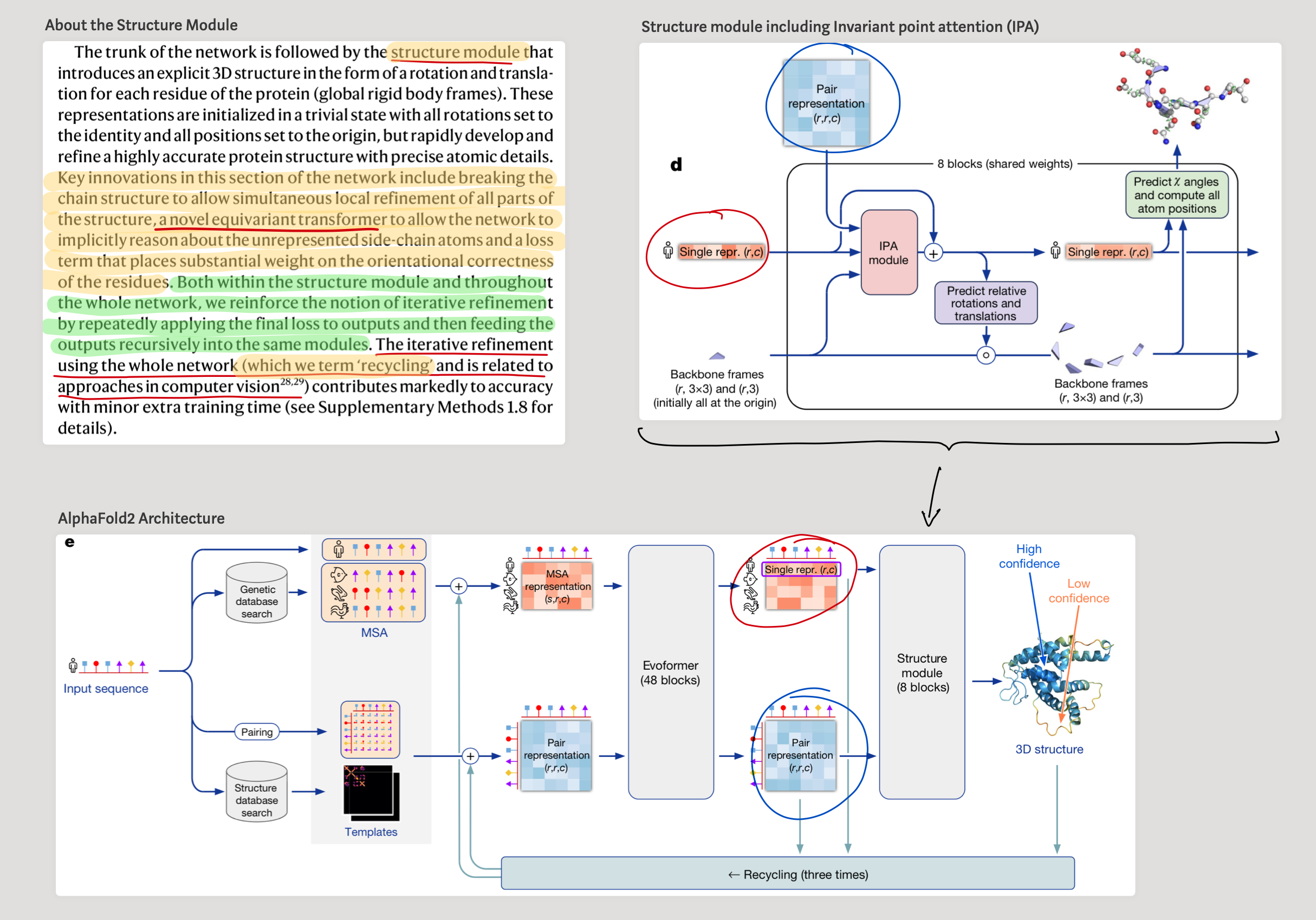
While authors extract the contact map from the attention maps, in order to extract spatial coordinates of the atoms, they use an equivariant transformer. This is the structure module introduced in AlphaFold. This equivariant transformer makes it possible to project out the spatial coordinates of the atoms just by using the internal language model representation. This architecture is referred to in the paper as ESMFold.
Steps in ESMFold
- Process sequence through ESM-2
- Pass representation learnt by ESM-2 to a series of folding blocks
2.1 - Each block sequentially updates a sequence representation and a pairwise representation - Pass the output to the structure module
- (repeat with 3 steps of recycling) (view code)
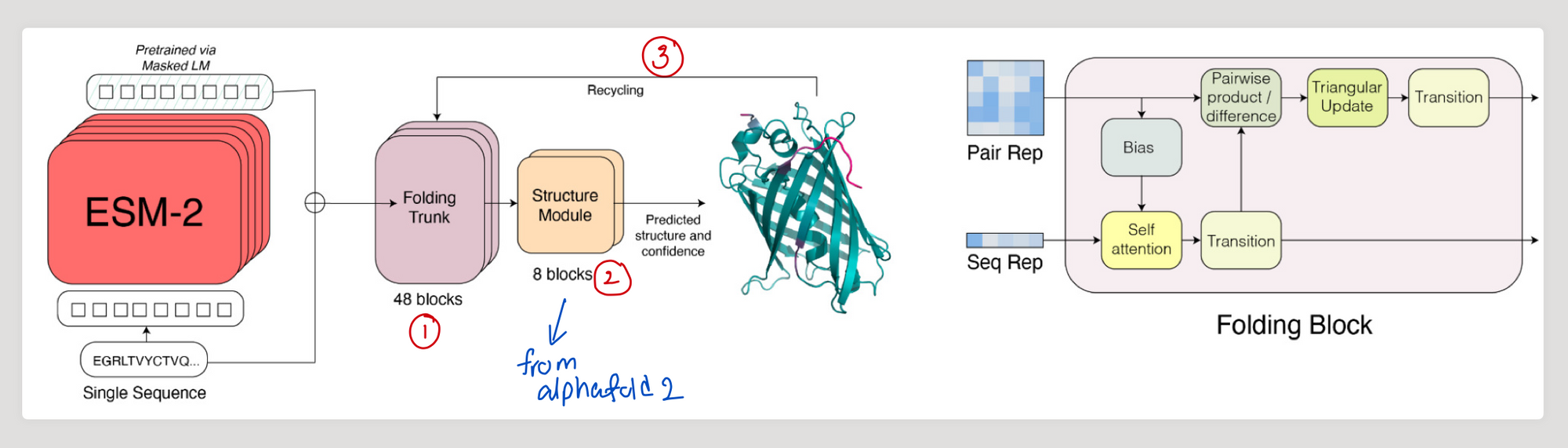

Training : To train the structure model to obtain spatial coordinates, they use, experimentally determined structures from PDB (~25K clusters covering a total of ~325K structures).
This is augmented with 12M structures predicted$^\ddagger$ with AlphaFold2.
Evaluation : 194 CAMEO Proteins and 51 CASP14 protiens
This language model based approach vastly simplifies the usual SOTA structure prediction process by eliminating the need for the following $^\dagger$,
- External evolutionary databases
- Multiple sequence alignments (MSAs)
- Templates
3. Results
3.1 - How well does it predict structures ?
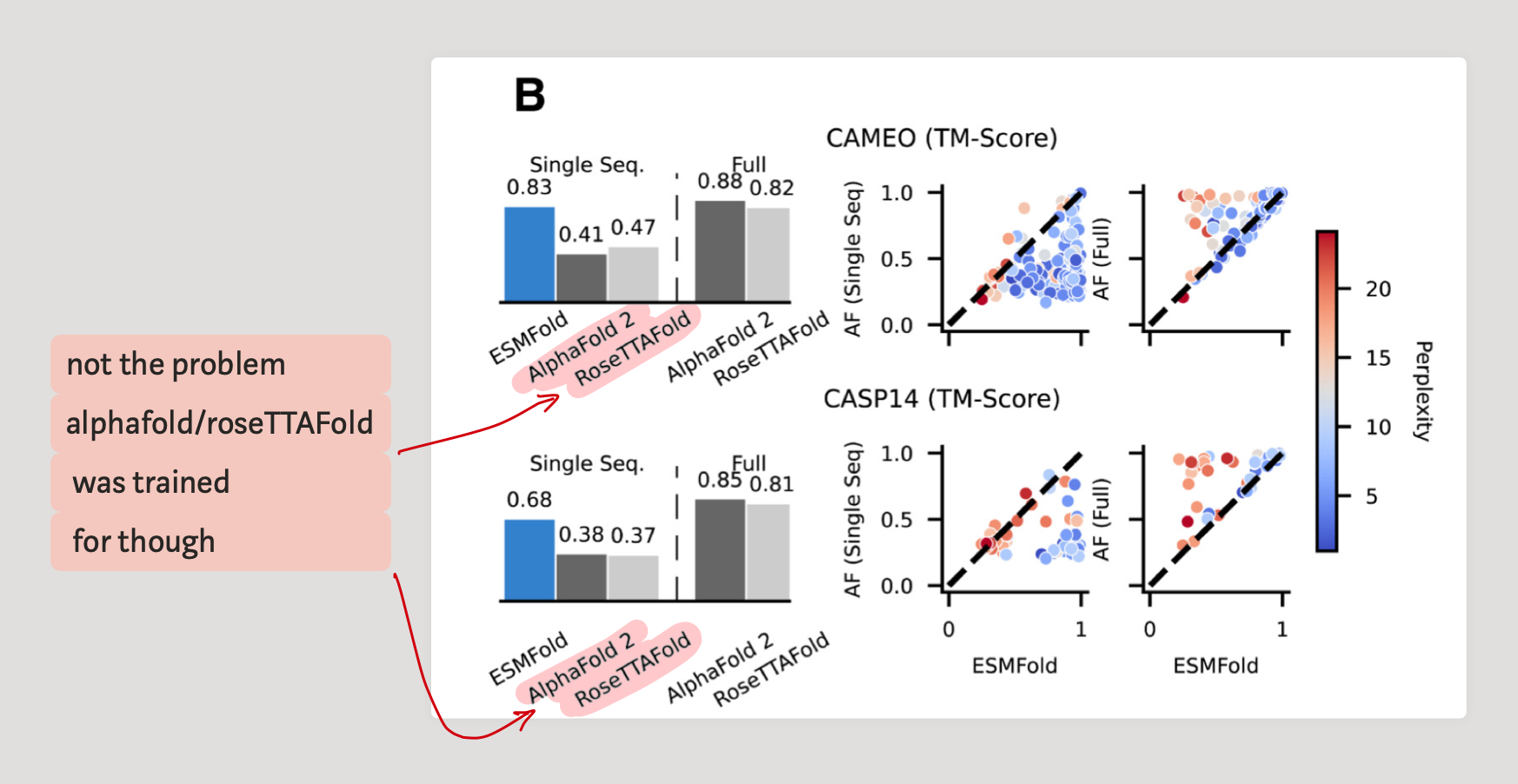
As mentioned before, they evaluate performance on CAMEO and CASP14 proteins and check how well the structure was predicted using the TM-Score.
In predicting the structure just by single sequences, ESMFold achieves very good performance compared to AlphaFold and RoseTTAFold.
3.2 - How important is the language model in the pipeline ?
The key question that arises is how important is the representation learnt by the LM for the task of structure prediction. To quantify this we need several metrics.
First, we need to characterize how good the understanding of the language model (ESM-2) is. This is where perplexity comes in. We already have the TM-score to determine how well a structure matches the groundtruth.
Thus, the graph to the right in Fig. 2B shows that,
- High ESMFold TM-Scores have low perplexity scores (numerically speaking, on CAMEO, Pearson correlation coefficient is -0.55 and in CASP14 it's -0.67)
How can we achieve better perplexity?
Okay, now we know that having better language model representation (lower perplexity) leads to better structure prediction.
So how can we achieve better language model representation ? 🤔 Is scaling all you need ?
To answer this question, authors explore the effect of scaling and look at what happens to the following :
- Precision @ L
- Change in perplexity
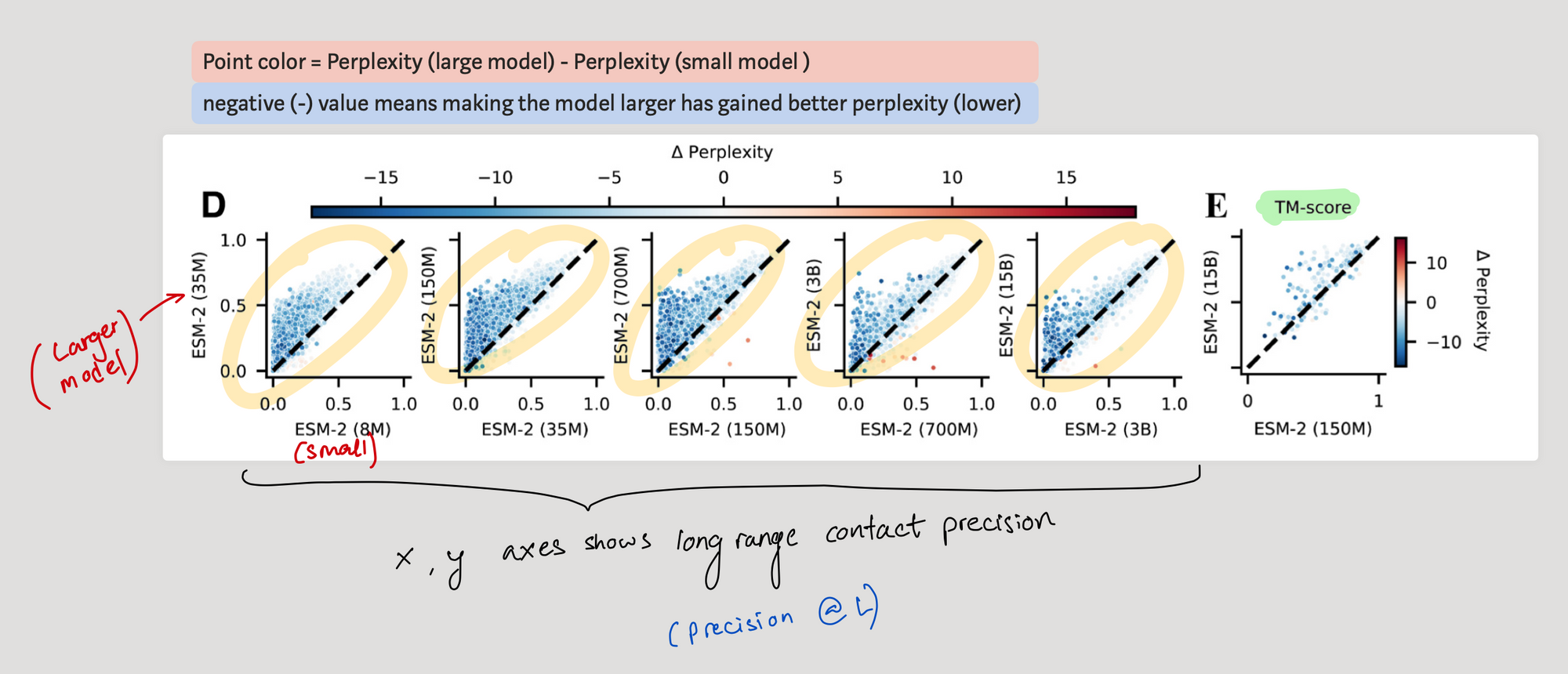
So they plot how the long range precision @ L changes once we move from a smaller model (x axis) to a larger one (y axis). From the points above the diagonal, it seems that scaling does help to achieve better long range precision @ L (some proteins show improvement).
So is scaling 'the' answer ?
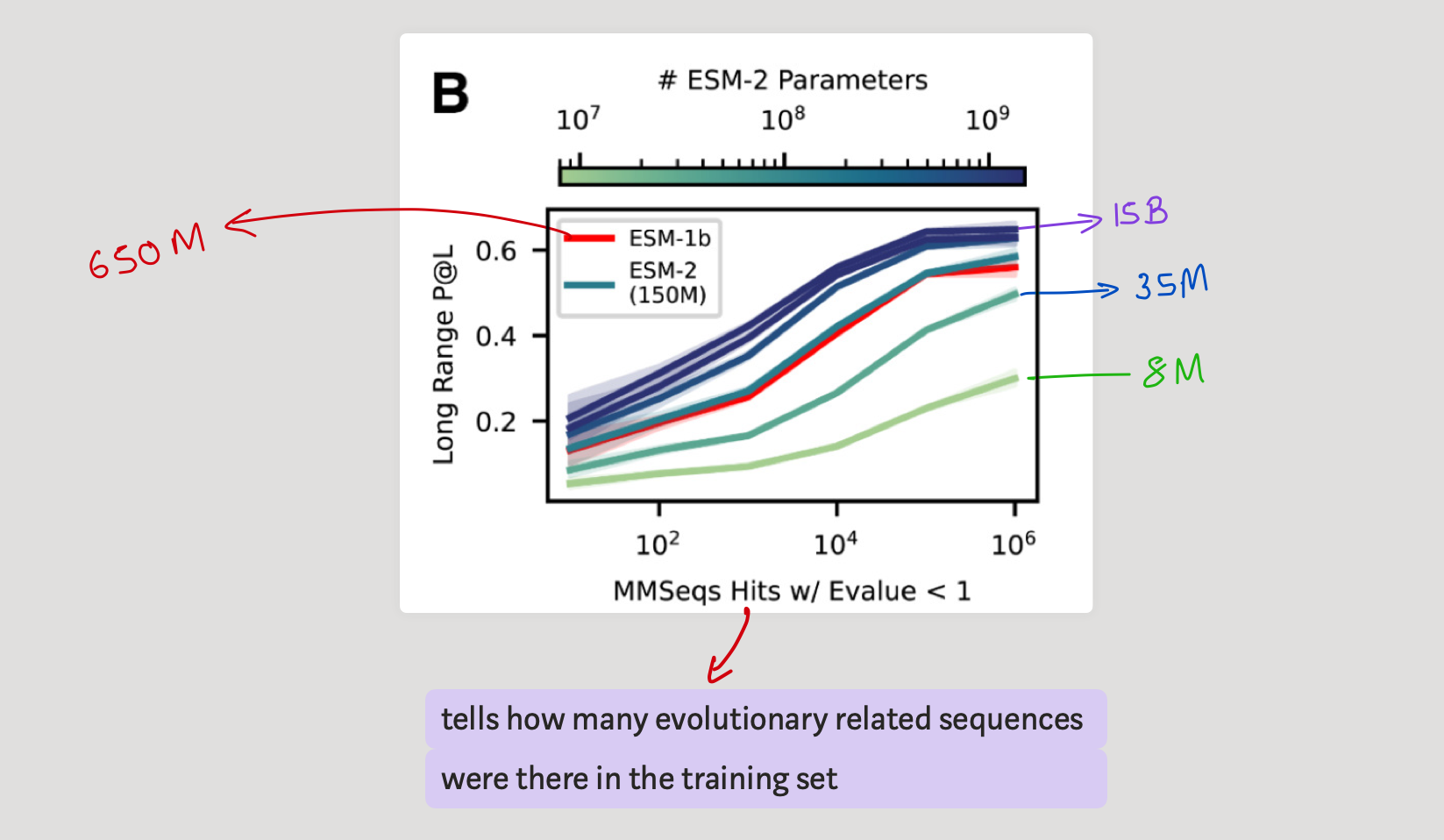
It seems it's not that simple. While certainly for some proteins P@L increases as we scale, if we look at the how many evolutionary related sequences were there, it tells another story. It seems that LMs cannot perform well when there is less number of relevant training data to the query we are asking. This seems intuitive though right? more studying more results? But the thing is it seems it's more memorization than understanding the subject?
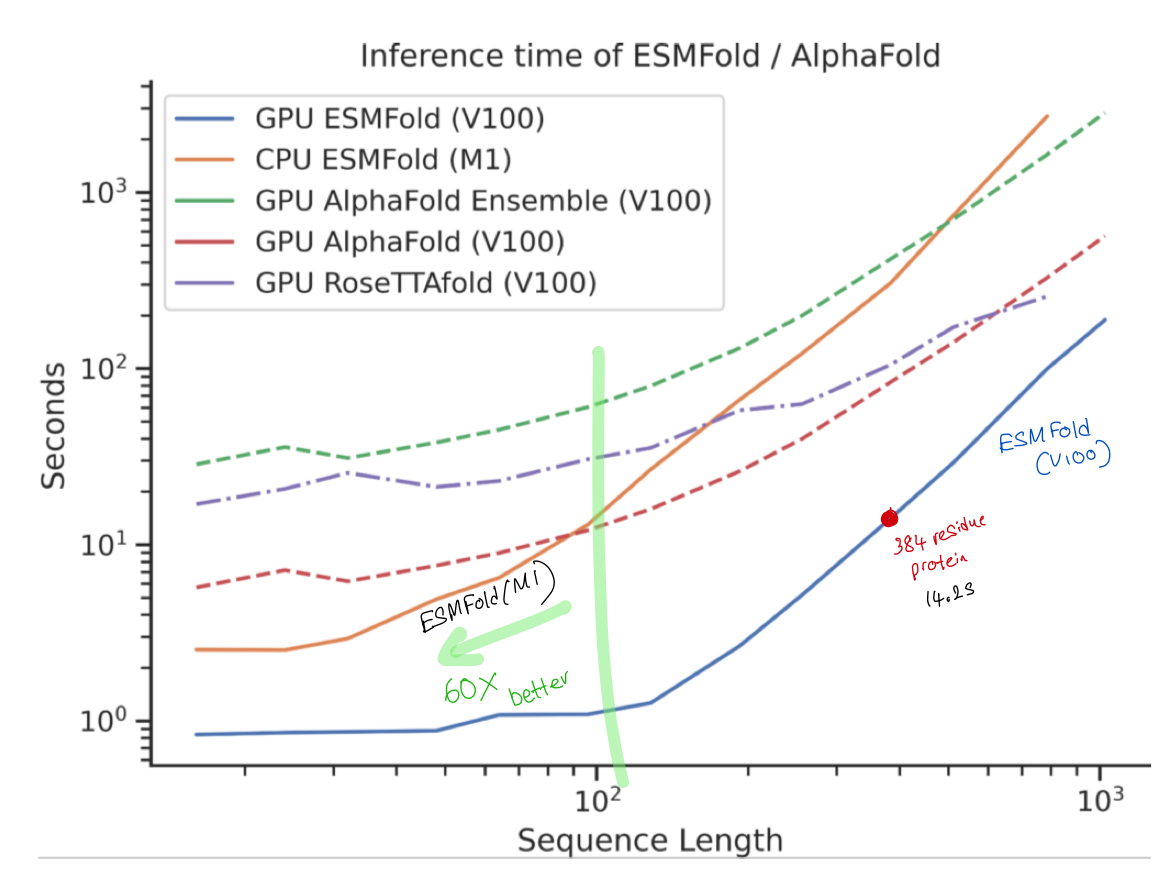
3.2 - Ok, what about prediction speed ?
- Protein with 384 residues on 1, NVIDIA V100 GPU => 14.2 Seconds$^\star$
- Shorter sequences ~60x speedup
3.3 - Comparison with other protein language models
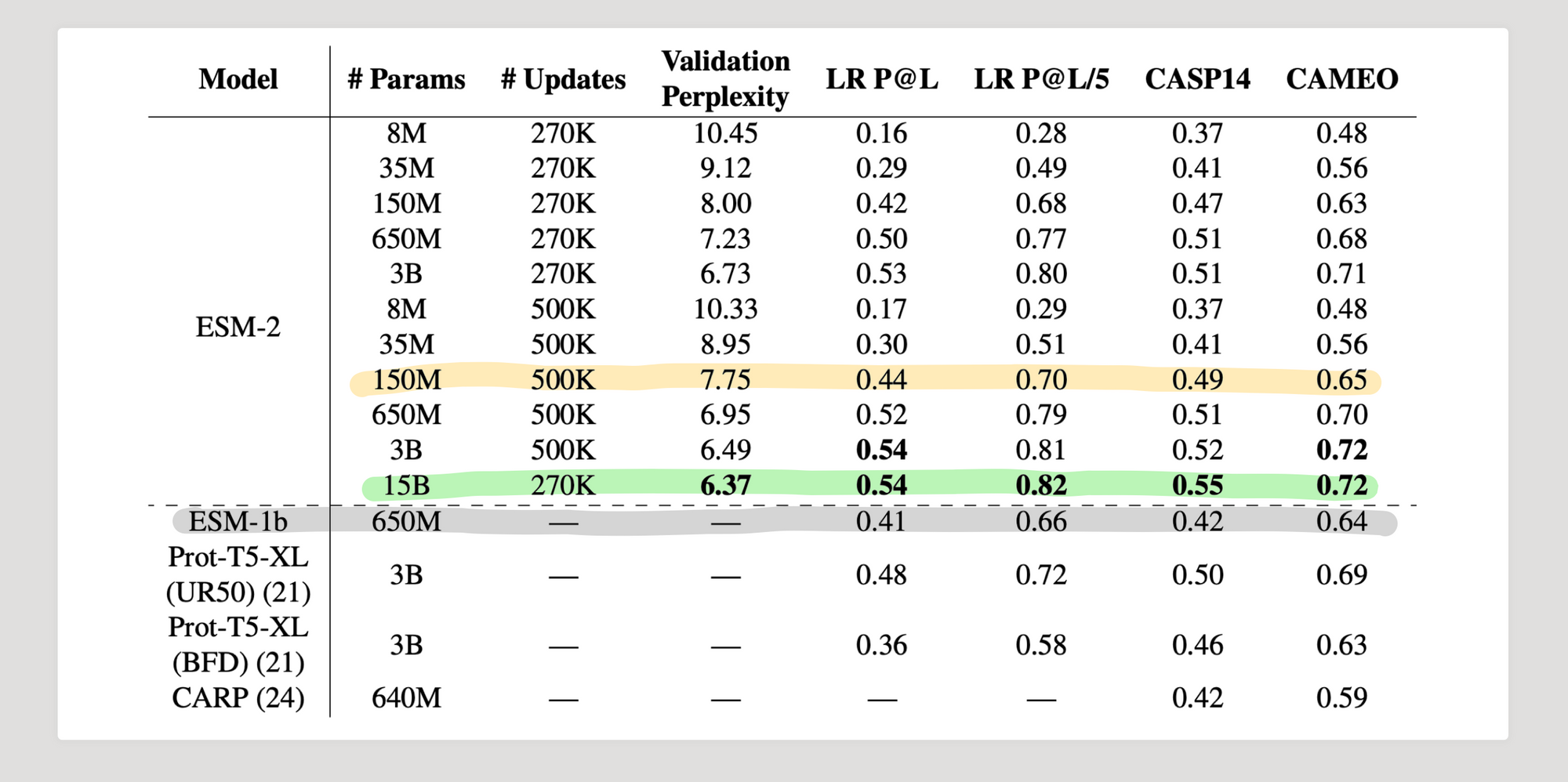
4. Conclusion
It's remarkable that these authors scale protein language models and it has resulted in learning structure hidden through databases of sequences, and thus we do not need to depend onto the MSA.
Is it because, the model has learnt to obtain the signal which we previously obtained through MSAs? What can we tell about the performance of sequences that had less number of evolutionary sequences in training data? why does it still struggle to obtain decent performance. It would be very interesting to analyze these directions.
Thanks for reading this, hope you found it useful. If you have any suggestions/ comments please share below.